SLC12A2 Related Disorders
NKCC1 Deficiency
SLC12A2 has recently been established as a disease causing gene. Back in 2015, the first documented human with an SLC12A2 mutation was found and extensive research followed leading to a better understanding of the Gene's function and the finding of more patients. It is now well established that the disruption of human SLC12A2 (NKCC1) may result in severe dysfunction involving multiple organs and systems. The spectrum of disorders caused by mutations in SLC12A2 is vast because the cotransporter is expressed in so many tissues. Our mission here is to connect others, educate, collaborate, and ultimately help find a cure or better treatment options for patients afflicted with SLC12A2- Related Disorders.
Please note: The information provided by The SLC12A2 Foundation is for educational purposes only. It should not be interpreted as a recommendation for a specific treatment plan, product, course of action, medical or healthcare provider. The information here does not replace medical consultations with a qualified professional to meet the individual’s needs. In addition, medical information can change rapidly; therefore, some information may be out of date, and/or contain inaccuracies or typographical errors. Ultimately, if you have concerns about your health, particularly SLC12A2-related disorders, you should consult your healthcare provider.)
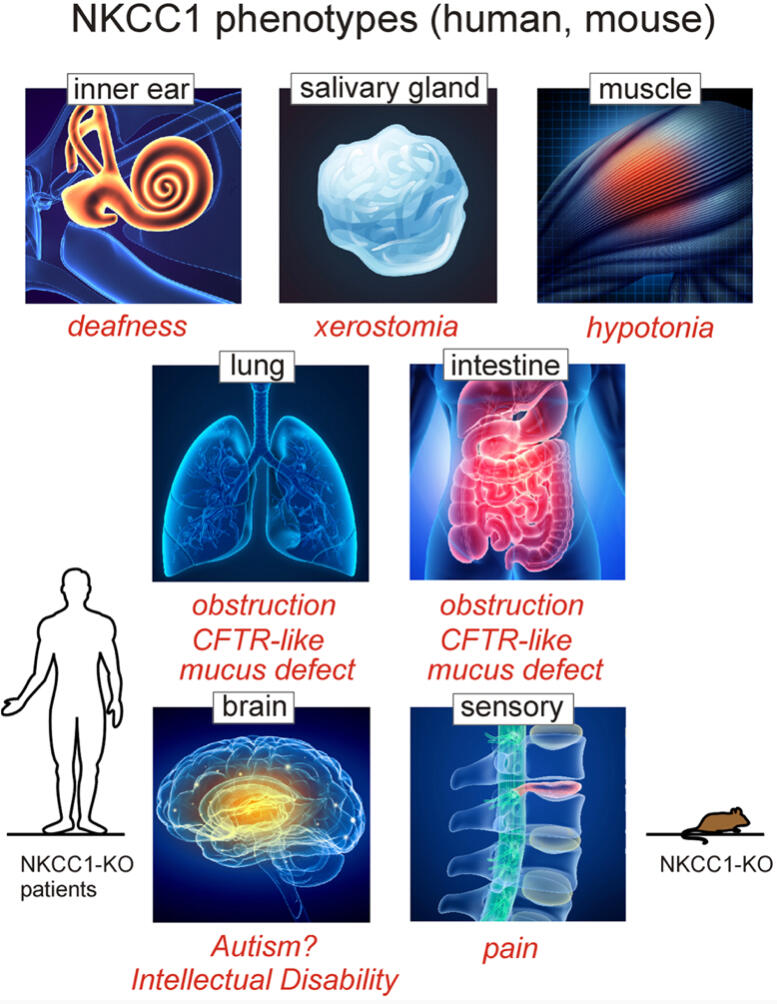
Early Cystic Fibrosis like presentation (usually in infancy) including failure to thrive, respiratory failure and mucos plugging episodes.Sensorineural hearing lossVestibular issuesMovement Disorders (myoclonus, fasciculations, episodic dystonia, parkinsonian-like movements, spastic dysplagia, spastic quadriparesis)Neuro-developmental Delays (including intellectual disabilities and autism spectrum)Hypotonia (low muscle tone)Muscle WeaknessExercise intoleranceAnhidrosis (sweating dysfunction)GI Tract dysfunction such as psuedo-obstruction, dysmotility, constipation, malrotation, GERD, episodic vomitting, dehydration, and intestinal obstructionDry Mouth and Salivary gland blockageEndocrine dysfunction (including low cortisol levels/adrenal gland dysfunction, panhypopituitarism, hypogonadism, hypothyroid, hypo-parathyroid, mineralocorticoid deficiency)Kidney Issues (renal tubular dysfunction, hypophosphatemia)Mitochondrial/ Metabolic DysfunctionOrthostatic IntoleranceEnlarged HeartEEG abnormalties (inconclusive)SeizuresNeurogenic Bladder
Patient Symptoms and Presentation
Patient symptoms in those found to have SLC12A2 mutations are vast due to the expression of the cotransporter in so many tissues. Like many novel genetic diseases, not all mutations necessarily cause dysfunction. There is so much yet to be discovered. This information is ever changing and therefore not exclusive. Updates will be done as new research and patient information comes in. Not all patients have these symptoms. Many have a few of them combined, some have only hearing loss, and the known patients with significant mutations seem to have multiple system dysfunction. It should be noted that not all SLC12A2 patients have neuro-developmental delays. At this time, there are multiple confirmed patients that are neuro-typical. In addition to this, while many of the patients have hearing loss, some have adequate hearing and should not be overlooked. Genetic testing should still be considered by physicians when one or more of these systems are affected with no known cause, or a patient presents with a Cystic Fibrosis type disorder in infancy with negative genetic and abnormal sweat tests.Currently there is no specific treatment options for SLC12A2 Related Disorders other than treating the symptoms and organ dysfunctions. Depending on involvement, patients may benefit from multiple speciality consultations, various therapies (including occupational, physical, and behavioral), cochlear implants when indicated, speech therapy, deaf and hard of hearing services, nutritional support, and/or mobility support when needed. Early intervention is imperative for the best outcomes and better quality of life.
Genetics and Testing
The SLC12A2 gene is located on the long arm of chromosome 5 at 5q23.3 (GRCh38.p13, 5:128083766-128189677). The gene is highly conserved among species and more than 32 orthologues have now been described. Human NKCC1 is composed of 27 exons, with two splice variants identified: NKCC1a (full length) and NKCC1b (lacking exon 21)SLC12A2 mutations are usually found with extensive whole genome testing. Thus far, confirmed patients with varying phenotypes have been found who exhibit all types of inheritance, including de nova.

PATIENT STORIES COMING SOON!




CURRENT PUBLISHED RESEARCH
NKCC1: Newly Found as a Human Disease-Causing Ion Transporter
ArticleSLC12A2 mutations cause NKCC1 deficiency with encephalopathy and impaired secretory epithelia
ArticleSLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect
ArticleA mutation in the Na-K-2Cl cotransporter-1 leads to changes in cellular metabolism
ArticleNovel Human NKCC1 Mutations Cause Defects in Goblet Cells Mucus Secretion and Chronic Inflammation
ArticleA dileucine motif in the COOH-terminal domain of NKCC1 targets the cotransporter to the plasma membrane.
ArticleKilquist syndrome: A novel syndromic hearing loss disorder caused by homozygous deletion of SLC12A2
ArticleMistargeting of a truncated Na-K-2Cl cotransporter in epithelial cells
ArticleA patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K2Cl cotransporter, NKCC1
[Article] (https://www.ncbi.nlm.nih.gov/.../pdf/DelpireMCS001289.pdf)Functional Characterization of the First Known Mutation of the Human SLC12A2 (NKCC1) Gene
Article

Contact
Please contact us below at the following email with any questions, comments or concerns related to SLC12A2 mutations and disorders.
Thank you
Thank you for your email. We will get back to you as soon as we can:)
Text Text Text TextText Text Text Text
Text Text Text Text
Text Text Text TextText Text Text Textvv
v
Text Text Text Text
Text Text Text Text